Ms.GSI in the multistage of genetic stock identification madness
Source:vignettes/msgsi_vignette.Rmd
msgsi_vignette.RmdOverview
This document contains the background information for a integrated multistage genetic stock identification (GSI) model in two parts. The first part the describes how to use package Ms.GSI to conduct a GSI analysis. The steps include formatting input data, running the integrated multistage GSI model, and summarizing results. The second part details the technical background of integrated multistage GSI model and its mathematical theory. There is a separate article describe the general background of the integrated multistage framework.
How to use Ms.GSI
Ms.GSI follows the work flow: format input data, run integrated multistage model, and summarize results/convergence diagnostics.
Input data
There are few pieces of information needed for input data set:
- broad-scale baseline
- regional baseline
- mixture sample
- broad-scale population information
- regional population information
There are pre-loaded example data sets available in Ms.GSI. We will look at them one at a time. Note that the example data sets are simulated using existing baseline archived by the Alaska Department of Fish & Game Gene Conservation Lab (GCL). The fabricated data set does not represent the true population proportions in real fisheries. In this example, we made up a scenario similar to the Bering Sea groundfish fisheries where Chinook salmon harvested as bycatch originated from a wide range of geographic areas. Within the bycatch sample, we were interested in the proportion contribution from the lower, middle and upper Yukon River reporting groups (see map below). We used a coast-wide data set for Chinook salmon (Templin et al. 2011; Templin baseline hereafter) as the broad-scale baseline to separate Yukon River fish from other non-Yukon stocks in our data set during the first stage of the analysis. However, genetic markers in the Templin baseline were unable to clearly distinguish between lower Yukon River and other coastal western Alaska populations, so we used a second baseline with additional genetic markers that were specifically designed to differentiate Yukon River Chinook salmon populations (Lee et al. 2021; Yukon River baseline hereafter) as the regional fine-scale baseline for the second stage.
It is important to note that in the original grouping of the Templin baseline, Lower Yukon was a part of Coastal Western Alaska reporting group. We isolated Lower Yukon from the rest of the Coastal Western Alaska, but the accuracy of the proportion estimates would likely diminish because of the breakup of Coastal Western Alaska reporting group. We do not recommend using a genetic baseline beyond its original design. And researchers should be aware of the capability of their genetic baselines before utilizing them in the integrated multistage model. In our example, it would be ideal to keep the Coastal Western Alaska group intact in the broad-scale baseline, and break up the group into Lower Yukon and others using a fine-scale regional baseline. However, at the time of writing, such regional baseline with adequate resolution was still in development.
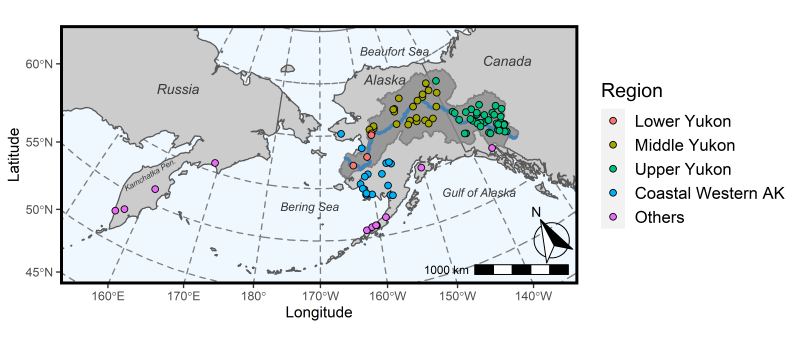
We assembled a mixture sample containing 150 individuals from collection sites across Yukon River, coastal western Alaskan, Alaska Peninsula, Gulf of Alaska, and Kamchatka Peninsula (Russia). The collections were grouped into five reporting groups: Lower Yukon, Middle Yukon, Upper Yukon, Coastal Western Alaska (Coastal West Alaska), and Others.
Mixture
First we will take a look at the baseline and mixture samples. Ms.GSI accepts the genotype information in two format: 1) GCL format or 2) package rubias format. The example data sets are in rubias format and naming convention, but procedures for GCL format are the same1.
print(dplyr::as_tibble(mix))
#> # A tibble: 150 × 360
#> sample_type repunit collection known_collection_t1 known_collection_t2 indiv
#> <chr> <lgl> <chr> <chr> <chr> <chr>
#> 1 mixture NA Bering Sea NA NA fish_1
#> 2 mixture NA Bering Sea NA NA fish_2
#> 3 mixture NA Bering Sea NA NA fish_3
#> 4 mixture NA Bering Sea NA NA fish_4
#> 5 mixture NA Bering Sea NA NA fish_5
#> 6 mixture NA Bering Sea NA NA fish_6
#> 7 mixture NA Bering Sea NA NA fish_7
#> 8 mixture NA Bering Sea NA NA fish_8
#> 9 mixture NA Bering Sea NA NA fish_9
#> 10 mixture NA Bering Sea NA NA fish_…
#> # ℹ 140 more rows
#> # ℹ 354 more variables: `GTH2B-550` <chr>, `GTH2B-550.1` <chr>, NOD1 <chr>,
#> # NOD1.1 <chr>, `Ots_100884-287` <chr>, `Ots_100884-287.1` <chr>,
#> # `Ots_101554-407` <chr>, `Ots_101554-407.1` <chr>, `Ots_102414-395` <chr>,
#> # `Ots_102414-395.1` <chr>, `Ots_102867-609` <chr>, `Ots_102867-609.1` <chr>,
#> # `Ots_103041-52` <chr>, `Ots_103041-52.1` <chr>, `Ots_103122-180` <chr>,
#> # `Ots_103122-180.1` <chr>, `Ots_104048-194` <chr>, …Columns 5 to 358 contain genotype information for loci in BOTH broad-scale and regional baselines. You do not have to specify the loci for each baseline (you can if you want to double check, more on that later). Ms.GSI matches the loci between mixture and baselines as long as the locus names are consistent.
If you have fish with known-origin, you can specify their identities
by adding columns called known_collection_t1 and
known_collection_t2 for the broad-scale and regional
baselines, respectively, in the mixture data set. The entry for
known-origin should match the collection name in the baseline. Fish with
unknown-origin should have a NA entry.
Broad-scale baseline
Next, we will take a look at the broad-scale (Templin) baseline example provided in Ms.GSI. There are originally 45 loci in the Templin baseline, but we reduced the marker set to 28 loci due to limitation on the data size (and other technical reasons). However, for demonstration purpose, this data set will suffice.
print(dplyr::as_tibble(base_templin))
#> # A tibble: 29,363 × 60
#> sample_type repunit collection indiv `GTH2B-550` `GTH2B-550.1` NOD1 NOD1.1
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 reference Russia KBIST98L KBIST9… C C C G
#> 2 reference Russia KBIST98L KBIST9… C G C G
#> 3 reference Russia KBIST98L KBIST9… C G C G
#> 4 reference Russia KBIST98L KBIST9… C C G G
#> 5 reference Russia KBIST98L KBIST9… C G G G
#> 6 reference Russia KBIST98L KBIST9… C C C C
#> 7 reference Russia KBIST98L KBIST9… C G C G
#> 8 reference Russia KBIST98L KBIST9… C C C G
#> 9 reference Russia KBIST98L KBIST9… C G C G
#> 10 reference Russia KBIST98L KBIST9… C C G G
#> # ℹ 29,353 more rows
#> # ℹ 52 more variables: `Ots_2KER-137` <chr>, `Ots_2KER-137.1` <chr>,
#> # `Ots_AsnRS-72` <chr>, `Ots_AsnRS-72.1` <chr>, Ots_ETIF1A <chr>,
#> # Ots_ETIF1A.1 <chr>, `Ots_GPH-318` <chr>, `Ots_GPH-318.1` <chr>,
#> # `Ots_GST-207` <chr>, `Ots_GST-207.1` <chr>, `Ots_hnRNPL-533` <chr>,
#> # `Ots_hnRNPL-533.1` <chr>, `Ots_HSP90B-100` <chr>, `Ots_HSP90B-100.1` <chr>,
#> # `Ots_IGF1-91` <chr>, `Ots_IGF1-91.1` <chr>, `Ots_IK1-328` <chr>, …Regional baseline
The regional baseline (Yukon) is in the same format. There are originally 380 loci in the Yukon River Chinook baseline, but we reduced the numbers to 177 in this demonstration.
print(dplyr::as_tibble(base_yukon))
#> # A tibble: 5,435 × 358
#> sample_type repunit collection indiv `GTH2B-550` `GTH2B-550.1` NOD1 NOD1.1
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 reference Lower Yu… KANDR02.K… KAND… G G C C
#> 2 reference Lower Yu… KANDR02.K… KAND… C G C C
#> 3 reference Lower Yu… KANDR02.K… KAND… G G C C
#> 4 reference Lower Yu… KANDR02.K… KAND… G G C G
#> 5 reference Lower Yu… KANDR02.K… KAND… C G C C
#> 6 reference Lower Yu… KANDR02.K… KAND… C C C C
#> 7 reference Lower Yu… KANDR02.K… KAND… C G G G
#> 8 reference Lower Yu… KANDR02.K… KAND… C C G G
#> 9 reference Lower Yu… KANDR02.K… KAND… C C G G
#> 10 reference Lower Yu… KANDR02.K… KAND… C G C G
#> # ℹ 5,425 more rows
#> # ℹ 350 more variables: `Ots_100884-287` <chr>, `Ots_100884-287.1` <chr>,
#> # `Ots_101554-407` <chr>, `Ots_101554-407.1` <chr>, `Ots_102414-395` <chr>,
#> # `Ots_102414-395.1` <chr>, `Ots_102867-609` <chr>, `Ots_102867-609.1` <chr>,
#> # `Ots_103041-52` <chr>, `Ots_103041-52.1` <chr>, `Ots_103122-180` <chr>,
#> # `Ots_103122-180.1` <chr>, `Ots_104048-194` <chr>, `Ots_104048-194.1` <chr>,
#> # `Ots_104063-132` <chr>, `Ots_104063-132.1` <chr>, `Ots_104415-88` <chr>, …Population information
Another piece of information needed is the population details for
each baseline. You need to include three columns in the population
information table. Column collection contains the names for
each population in the baseline. Column reunit specifies
the reporting group each population belongs to. Column
grpvec specifies the identification number for each
reporting group. Below shows the first ten rows of population
information for the Templin (broad-scale) baseline.
print(dplyr::as_tibble(templin_pops211))
#> # A tibble: 211 × 3
#> collection repunit grpvec
#> <chr> <chr> <dbl>
#> 1 KBIST98L Russia 1
#> 2 KBOLS02.KBOLS98E Russia 1
#> 3 KKAMC97.KKAMC98L Russia 1
#> 4 KPAKH02 Russia 1
#> 5 KPILG05.KPILG06 Coastal West Alaska 2
#> 6 KUNAL05 Coastal West Alaska 2
#> 7 KGOLS05.KGOLS06 Coastal West Alaska 2
#> 8 KANDR02.KANDR03 Lower Yukon 3
#> 9 KANVI02 Lower Yukon 3
#> 10 KGISA01 Lower Yukon 3
#> # ℹ 201 more rowsIf you have hatchery populations in your mixture sample, you can tell
Ms.GSI either a collection belongs to natural or
hatchery-origin by adding an origin column in the
population information table. In the origin column, you
identify each collection with "wild" or
"hatchery". If you don’t care to separate natural and
hatchery origins, you can lump them in one collection. In this case, you
don’t need to add an origin column. Also, this option to
identify hatchery fish is only available in the broad-scale baseline, so
don’t add an origin column in the population table for
regional baseline.
Population information table for the Yukon (regional) baseline is in the same format, but not necessarily in the same order.
print(dplyr::as_tibble(yukon_pops50))
#> # A tibble: 50 × 3
#> collection grpvec repunit
#> <chr> <dbl> <chr>
#> 1 KANDR02.KANDR03 1 Lower Yukon
#> 2 KANVI03.KANVI07 1 Lower Yukon
#> 3 KNUL12SF 1 Lower Yukon
#> 4 KNUL12NF 1 Lower Yukon
#> 5 KGISA01 1 Lower Yukon
#> 6 KKATE02.KKATE12 1 Lower Yukon
#> 7 KHENS01 2 Middle Yukon
#> 8 KHENS07.KHENS15 2 Middle Yukon
#> 9 KSFKOY03 2 Middle Yukon
#> 10 KMFKOY10.KMFKOY11.KMFKOY12.KMFKOY13 2 Middle Yukon
#> # ℹ 40 more rowsOnce you have the data files ready (I recommend saving them as .Rdata
or .Rds files), you can use prep_msgsi_data() function to
convert them into input data set for model run. You’ll also need to
identify “groups of interest” in parameter sub-group. In
this example, groups of interests are Lower Yukon, Middle Yukon and
Upper Yukon reporting groups. Their identify numbers are 3, 4, and 5 in
the broad-scale baseline. There’s an option to save the input data at a
designated directory by identifying the location in parameter
file_path.
If you have harvest information (or fishing effort), you should input
them using harvest_mean and harvest_cv
arguments. harvest_cv can be left without input if harvest
was estimated without errors. If no inputs here for harvest, we will
assume sample consists 100% of the harvest. Here we just made up a fake
total catch of 500 fish with CV of 0.05.
prep_msgsi_data() function matches the loci between
mixture and baselines. But if you want to make sure that you didn’t miss
any locus in your baselines or mixture, you can manually provide loci
names (in string vector) for each baseline by inputting them in
loci1 and loci2. In this example we don’t
manually provide lists of loci because we trust that mixture and
baselines all have the correct loci.
msgsi_dat <-
prep_msgsi_data(mixture_data = mix,
baseline1_data = base_templin, baseline2_data = base_yukon,
pop1_info = templin_pops211, pop2_info = yukon_pops50, sub_group = 3:5,
harvest_mean = 500, harvest_cv = 0.05)
#> Compiling input data, may take a minute or two...
#> Time difference of 10.55765 secsprep_msgsi_data() formats the data files and put them in
a list. It took few seconds to format the input data in this case.
Bigger data sets may take longer. Here are the first few rows/items in
the input data list:
lapply(msgsi_dat, head)
#> $x
#> # A tibble: 6 × 57
#> indiv `GTH2B-550_1` `GTH2B-550_2` NOD1_1 NOD1_2 `Ots_2KER-137_1`
#> <chr> <int> <int> <int> <int> <int>
#> 1 fish_1 1 1 2 0 0
#> 2 fish_2 2 0 1 1 0
#> 3 fish_3 2 0 1 1 0
#> 4 fish_4 2 0 2 0 0
#> 5 fish_5 1 1 1 1 0
#> 6 fish_6 2 0 1 1 0
#> # ℹ 51 more variables: `Ots_2KER-137_2` <int>, `Ots_AsnRS-72_1` <int>,
#> # `Ots_AsnRS-72_2` <int>, Ots_ETIF1A_1 <int>, Ots_ETIF1A_2 <int>,
#> # `Ots_GPH-318_1` <int>, `Ots_GPH-318_2` <int>, `Ots_GST-207_1` <int>,
#> # `Ots_GST-207_2` <int>, `Ots_HSP90B-100_1` <int>, `Ots_HSP90B-100_2` <int>,
#> # `Ots_IGF1-91_1` <int>, `Ots_IGF1-91_2` <int>, `Ots_IK1-328_1` <int>,
#> # `Ots_IK1-328_2` <int>, `Ots_LEI-292_1` <int>, `Ots_LEI-292_2` <int>,
#> # Ots_MHC1_1 <int>, Ots_MHC1_2 <int>, `Ots_OPLW-173_1` <int>, …
#>
#> $x2
#> # A tibble: 6 × 355
#> indiv `GTH2B-550_1` `GTH2B-550_2` NOD1_1 NOD1_2 `Ots_100884-287_1`
#> <chr> <int> <int> <int> <int> <int>
#> 1 fish_1 1 1 2 0 2
#> 2 fish_2 0 2 1 1 2
#> 3 fish_3 0 2 1 1 1
#> 4 fish_4 0 2 2 0 1
#> 5 fish_5 1 1 1 1 2
#> 6 fish_6 0 2 1 1 1
#> # ℹ 349 more variables: `Ots_100884-287_2` <int>, `Ots_101554-407_1` <int>,
#> # `Ots_101554-407_2` <int>, `Ots_102414-395_1` <int>,
#> # `Ots_102414-395_2` <int>, `Ots_102867-609_1` <int>,
#> # `Ots_102867-609_2` <int>, `Ots_103041-52_1` <int>, `Ots_103041-52_2` <int>,
#> # `Ots_103122-180_1` <int>, `Ots_103122-180_2` <int>,
#> # `Ots_104048-194_1` <int>, `Ots_104048-194_2` <int>,
#> # `Ots_104063-132_1` <int>, `Ots_104063-132_2` <int>, …
#>
#> $y
#> # A tibble: 6 × 59
#> collection repunit grpvec `GTH2B-550_1` `GTH2B-550_2` NOD1_1 NOD1_2
#> <chr> <chr> <dbl> <int> <int> <int> <int>
#> 1 CHBIG92.KIBIG93.KBIG… Northe… 9 254 82 104 228
#> 2 CHCRY92.KICRY94.KCRY… Coasta… 10 306 302 120 488
#> 3 CHDMT92.KDEER94 Coasta… 10 178 116 77 217
#> 4 CHKAN92.KIKAN93.KKAN… Coasta… 2 341 147 281 199
#> 5 CHKOG92.KIKOG93.KKOG… Coasta… 2 205 91 191 105
#> 6 CHNUU92.KINUS93 Coasta… 2 85 27 73 41
#> # ℹ 52 more variables: `Ots_2KER-137_1` <int>, `Ots_2KER-137_2` <int>,
#> # `Ots_AsnRS-72_1` <int>, `Ots_AsnRS-72_2` <int>, Ots_ETIF1A_1 <int>,
#> # Ots_ETIF1A_2 <int>, `Ots_GPH-318_1` <int>, `Ots_GPH-318_2` <int>,
#> # `Ots_GST-207_1` <int>, `Ots_GST-207_2` <int>, `Ots_HSP90B-100_1` <int>,
#> # `Ots_HSP90B-100_2` <int>, `Ots_IGF1-91_1` <int>, `Ots_IGF1-91_2` <int>,
#> # `Ots_IK1-328_1` <int>, `Ots_IK1-328_2` <int>, `Ots_LEI-292_1` <int>,
#> # `Ots_LEI-292_2` <int>, Ots_MHC1_1 <int>, Ots_MHC1_2 <int>, …
#>
#> $y2
#> # A tibble: 6 × 357
#> collection grpvec repunit `GTH2B-550_1` `GTH2B-550_2` NOD1_1 NOD1_2
#> <chr> <dbl> <chr> <int> <int> <int> <int>
#> 1 CHSID92j 3 Upper … 7 183 116 74
#> 2 K100MILECR16.K100MIL… 3 Upper … 7 103 78 34
#> 3 KANDR02.KANDR03 1 Lower … 78 230 208 100
#> 4 KANVI03.KANVI07 1 Lower … 62 164 131 79
#> 5 KBEAV97 2 Middle… 40 148 152 38
#> 6 KBIGS87.KBIGS07 3 Upper … 30 258 231 65
#> # ℹ 350 more variables: `Ots_100884-287_1` <int>, `Ots_100884-287_2` <int>,
#> # `Ots_101554-407_1` <int>, `Ots_101554-407_2` <int>,
#> # `Ots_102414-395_1` <int>, `Ots_102414-395_2` <int>,
#> # `Ots_102867-609_1` <int>, `Ots_102867-609_2` <int>,
#> # `Ots_103041-52_1` <int>, `Ots_103041-52_2` <int>, `Ots_103122-180_1` <int>,
#> # `Ots_103122-180_2` <int>, `Ots_104048-194_1` <int>,
#> # `Ots_104048-194_2` <int>, `Ots_104063-132_1` <int>, …
#>
#> $iden
#> # A tibble: 6 × 3
#> indiv id1 id2
#> <chr> <chr> <chr>
#> 1 fish_1 NA NA
#> 2 fish_2 NA NA
#> 3 fish_3 NA NA
#> 4 fish_4 NA NA
#> 5 fish_5 NA NA
#> 6 fish_6 NA NA
#>
#> $nalleles
#> GTH2B-550 NOD1 Ots_2KER-137 Ots_AsnRS-72 Ots_ETIF1A Ots_GPH-318
#> 2 2 2 2 2 2
#>
#> $nalleles2
#> GTH2B-550 NOD1 Ots_100884-287 Ots_101554-407 Ots_102414-395
#> 2 2 2 2 2
#> Ots_102867-609
#> 2
#>
#> $groups_t1
#> # A tibble: 6 × 3
#> collection repunit grpvec
#> <chr> <chr> <dbl>
#> 1 CHBIG92.KIBIG93.KBIGB04.KBIGB95 Northeast Gulf of Alaska 9
#> 2 CHCRY92.KICRY94.KCRYA05 Coastal Southeast Alaska 10
#> 3 CHDMT92.KDEER94 Coastal Southeast Alaska 10
#> 4 CHKAN92.KIKAN93.KKANE05 Coastal West Alaska 2
#> 5 CHKOG92.KIKOG93.KKOGR05 Coastal West Alaska 2
#> 6 CHNUU92.KINUS93 Coastal West Alaska 2
#>
#> $groups_t2
#> # A tibble: 6 × 3
#> collection repunit grpvec
#> <chr> <chr> <dbl>
#> 1 CHSID92j Upper Yukon 3
#> 2 K100MILECR16.K100MILECR15 Upper Yukon 3
#> 3 KANDR02.KANDR03 Lower Yukon 1
#> 4 KANVI03.KANVI07 Lower Yukon 1
#> 5 KBEAV97 Middle Yukon 2
#> 6 KBIGS87.KBIGS07 Upper Yukon 3
#>
#> $comb_groups
#> # A tibble: 6 × 2
#> collection repunit
#> <chr> <chr>
#> 1 CHBIG92.KIBIG93.KBIGB04.KBIGB95 Northeast Gulf of Alaska
#> 2 CHCRY92.KICRY94.KCRYA05 Coastal Southeast Alaska
#> 3 CHDMT92.KDEER94 Coastal Southeast Alaska
#> 4 CHKAN92.KIKAN93.KKANE05 Coastal West Alaska
#> 5 CHKOG92.KIKOG93.KKOGR05 Coastal West Alaska
#> 6 CHNUU92.KINUS93 Coastal West Alaska
#>
#> $sub_group
#> [1] 3 4 5
#>
#> $group_names_t1
#> [1] "Russia" "Coastal West Alaska" "Lower Yukon"
#> [4] "Middle Yukon" "Upper Yukon" "North Alaska Peninsula"
#>
#> $group_names_t2
#> [1] "Lower Yukon" "Middle Yukon" "Upper Yukon"
#>
#> $wildpops
#> [1] "CHBIG92.KIBIG93.KBIGB04.KBIGB95" "CHCRY92.KICRY94.KCRYA05"
#> [3] "CHDMT92.KDEER94" "CHKAN92.KIKAN93.KKANE05"
#> [5] "CHKOG92.KIKOG93.KKOGR05" "CHNUU92.KINUS93"
#>
#> $hatcheries
#> NULL
#>
#> $harvest
#> [1] 465.6346 505.7880 442.1161 499.2372 515.1293 528.8709Genetic stock identification
Once your input data set is ready, you can use
msgsi_mdl() to run the model. If you are used to running
rubias, Ms.GSI might feel a bit slow. That is because
1) we are running two GSI models in tandem, so it takes twice as long
than running a single model, and 2) Ms.GSI is written solely in
R, which is not as computationally efficient as language
C. So, why not code Ms.GSI in C? Because
we’re not technically advanced like the folks who developed
rubias package (i.e., we don’t know how to code in
C++).
Because of the running time, we recommend running the integrated multistage model in conditional GSI mode (default setting). But there is an option to run the model in fully Bayesian mode if one choose to. If you run the model in fully Bayesian mode, you have the option to include numbers of adaptation run. Some people think that adaptation run encourages convergence in fully Bayesian mode. We have not test that theory but provide the option for those who want to try it.
We demonstrate the model run with one chains of 150 iterations (first
50 as warm-up runs, or burn-ins). We only run one chain in this example
so it can pass CMD check while building the vignette document2. In
reality, you should definitely run multiple chains with
more iterations. There are also options to keep the burn-ins and set
random seed for reproducible results. We don’t show them in this example
though (but you can always ?msgsi_mdl).
msgsi_out <- msgsi_mdl(msgsi_dat, nreps = 150, nburn = 50, thin = 1, nchains = 1)
#> Running model... and good things come to Femme Queen Vogue!
#> Time difference of 3.196053 secs
#> July-02-2026 19:00Summarizing results
Stock proportions
The output of model contains nine items: summ_t1,
trace_t1, summ_t2, trace_t2,
summ_comb, trace_comb,
comb_groups, comb_groups, iden_t1
and idens_t2. Items with “summ” are summary for reporting
group proportions and associated convergence diagnostics. If you want to
see summaries for stage one and two individually, summ_t1
and summ_t2 will show you that. Most people probably want
to see the combined summary, summ_comb.
msgsi_out$summ_comb
#> # A tibble: 12 × 10
#> group mean median sd ci.05 ci.95 p0 z0 GR n_eff
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <lgl> <dbl>
#> 1 Russia 5.28e-2 5.15e-2 0.0201 2.40e- 2 0.0882 0 0 NA 54.4
#> 2 Coastal We… 7.89e-2 7.89e-2 0.0720 5.68e-11 0.217 0.23 0.23 NA 6.31
#> 3 North Alas… 3.00e-2 2.38e-2 0.0307 2.15e-12 0.0952 0.28 0.26 NA 9.12
#> 4 Northwest … 2.81e-1 2.77e-1 0.0553 2.01e- 1 0.367 0 0 NA 26.9
#> 5 Copper 5.86e-4 2.89e-6 0.00153 9.26e-20 0.00475 0.86 0.88 NA 100
#> 6 Northeast … 2.75e-3 2.24e-5 0.00643 4.71e-18 0.0150 0.72 0.76 NA 15.1
#> 7 Coastal So… 7.18e-4 2.04e-7 0.00174 5.07e-18 0.00453 0.8 0.86 NA 100
#> 8 British Co… 1.20e-3 2.11e-6 0.00344 4.02e-21 0.00714 0.82 0.85 NA 41.9
#> 9 WA/OR/CA 4.15e-4 7.78e-7 0.00142 5.66e-15 0.00200 0.88 0.94 NA 100
#> 10 Lower Yukon 2.87e-1 2.96e-1 0.0905 1.25e- 1 0.415 0 0 NA 7.05
#> 11 Middle Yuk… 7.57e-2 7.41e-2 0.0230 4.52e- 2 0.118 0 0 NA 100
#> 12 Upper Yukon 1.89e-1 1.86e-1 0.0337 1.42e- 1 0.252 0 0 NA 100Most column names are self explanatory, but others might need some
additional descriptions. ci.05 and ci.95 are
the lower and upper bounds of 90% credible interval. p0 is
the probability of an estimate equals zero. We estimate p0
by calculating the portion of posterior samples that is less than
,
or 0.5/total harvest if harvest information is provided. GR
is the Gelman-Rubin statistic (a.k.a.
).
In this example, Gelman-Rubin statistic is not calculated because we
only run one chain. n_eff is the effective size, or
.
We will not discuss how to diagnose convergence in this document. Please
consult Gelman et al. 2014, Gelman & Rubin 1992, Brooks & Gelman
1998 and other literature on statistical methods. z0 is the
probability of an estimate equals zero based on history of individuals
assigned to each collection and reporting groups. Details of the theory
and calculation can be found here.
Items with “trace” are the posterior sample history, or trace
history, for either stage one, two, or combined. Trace history is needed
for making trace plots. And if you need to combine reporting groups
proportions or combine variance, trace histories are what you need.
trace_ items are tibbles with each collection as a column.
There are two additional columns, itr and
chain, to identify Markov chain Monte Carlo (MCMC) sampling
iteration and chain.
comb_groups are provided in the output as reference for
functions making trace plots (tr_plot()) and stratified
estimator (stratified_estimator_msgsi()). Grouping for
stage one and two can also be found in the input data.
msgsi_out$trace_comb
#> # A tibble: 100 × 238
#> CHBIG92.KIBIG93.KBIGB04.KBIGB95 CHCRY92.KICRY94.KCRYA05 CHDMT92.KDEER94
#> <dbl> <dbl> <dbl>
#> 1 6.54e-12 2.92e- 27 5.34e-298
#> 2 1.17e-55 7.27e- 16 3.43e- 51
#> 3 2.08e- 9 1.04e-158 1.24e-232
#> 4 5.32e-10 2.31e-232 2.79e- 12
#> 5 2.59e-26 3.46e- 96 2.08e- 78
#> 6 2.22e-38 1.11e- 65 2.23e-308
#> 7 1.04e- 8 3.59e-214 1.18e- 63
#> 8 1.00e-26 7.47e- 88 5.57e- 91
#> 9 3.33e-11 3.36e-242 8.72e- 41
#> 10 1.85e- 6 2.23e-308 1.49e- 41
#> # ℹ 90 more rows
#> # ℹ 235 more variables: CHKAN92.KIKAN93.KKANE05 <dbl>,
#> # CHKOG92.KIKOG93.KKOGR05 <dbl>, CHNUU92.KINUS93 <dbl>,
#> # CHTAH92.KTAHI04 <dbl>, CHWHI92.KWHIT98.KWHITC05 <dbl>, KALSE04 <dbl>,
#> # KANCH06.KANCH10 <dbl>, KANDR89.KANDR04 <dbl>, KAROL05 <dbl>,
#> # KBENJ05.KBENJ06 <dbl>, KBIGCK04 <dbl>, KBIGQU96 <dbl>,
#> # KBIRK01.KBIRK02.KBIRK03.KBIRK97.KBIRK99 <dbl>, KBIST98L <dbl>, …Stock-specific total catch
The output also includes the trace history of harvest count for each
reporting group. The output is in the long-form format of tidyverse;
there are three columns (ac, pprc, and sstc)
that represent fishery harvest. Following the same vocabulary as the
rubias package, ac column represents assignment
counts and is summarized based on individual assignments of the mixture
samples to each collection. pprc represents posterior
predictive remaining counts and is summarized based on Monte Carlo
simulations of the individual assignments of the un-sampled harvest. And
sstc represent total stock specific counts and is the sum of
ac and pprc. Other columns identify the collection,
reporting unit, MCMC chain and iteration of each harvest count.
msgsi_out$sstc_trace_t2
#> # A tibble: 5,000 × 8
#> ac pprc sstc collection itr ch repunit grpvec
#> <int> <int> <int> <chr> <dbl> <int> <chr> <dbl>
#> 1 0 0 0 CHSID92j 51 1 Upper Yukon 3
#> 2 0 0 0 K100MILECR16.K100MILECR15 51 1 Upper Yukon 3
#> 3 0 0 0 KANDR02.KANDR03 51 1 Lower Yukon 1
#> 4 6 9 15 KANVI03.KANVI07 51 1 Lower Yukon 1
#> 5 0 0 0 KBEAV97 51 1 Middle Yukon 2
#> 6 0 0 0 KBIGS87.KBIGS07 51 1 Upper Yukon 3
#> 7 0 0 0 KBLIN03.KBLIN08 51 1 Upper Yukon 3
#> 8 0 0 0 KCHAN04 51 1 Middle Yukon 2
#> 9 0 0 0 KCHAT01.KCHAT07 51 1 Middle Yukon 2
#> 10 0 0 0 KCHAU01.KCHAU03 51 1 Upper Yukon 3
#> # ℹ 4,990 more rowsStock-specific total catch and proportions of the reporting groups
for the combined baselines can be summarized using
stratified_estimator_msgsi() function (more on the
functionality of stratified_estimator_msgsi() later…). Here
the stock proportions and their associated uncertainty (sd, ci05, and
ci95) are calculated based on the trace history of the catch.
stratified_estimator_msgsi(msgsi_out, mixvec = "Bering example")
#> # A tibble: 12 × 15
#> repunit mean_sstc sd_sstc median_sstc ci05_sstc ci95_sstc mean sd
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 Northeast … 1.35 3.47 0 0 9 2.64e-3 0.00678
#> 2 Coastal So… 0.32 0.973 0 0 3 6.28e-4 0.00192
#> 3 Coastal We… 40.5 36.1 38 0 101. 8.10e-2 0.0716
#> 4 WA/OR/CA 0.13 0.630 0 0 1 2.58e-4 0.00126
#> 5 Northwest … 140. 25.6 139 101 175. 2.79e-1 0.0476
#> 6 British Co… 0.58 1.68 0 0 5 1.17e-3 0.00335
#> 7 Russia 26.4 8.88 26 12 40.1 5.28e-2 0.0174
#> 8 North Alas… 14.7 14.1 12 0 42.0 2.95e-2 0.0281
#> 9 Copper 0.29 1.01 0 0 2 5.91e-4 0.00204
#> 10 Upper Yukon 94.6 14.7 94 73 121. 1.89e-1 0.0282
#> 11 Lower Yukon 145. 46.6 151 66.8 209. 2.89e-1 0.0910
#> 12 Middle Yuk… 37.1 10.5 36.5 22 55.0 7.40e-2 0.0202
#> # ℹ 7 more variables: median <dbl>, ci05 <dbl>, ci95 <dbl>, `P=0` <dbl>,
#> # `Z=0` <dbl>, GR <lgl>, n_eff <dbl>Individual assignments
The next two items in the output are the identity assignment history of each individual in the mixture sample. Each column represents an individual in the mixture, and each row records the identity assigned during each iteration in each chain. These numbers are the population identifiers in the same order as your population information files (and baseline files). Individuals are ordered in the same as the input data (i.e., mixture data).
msgsi_out$idens_t1
#> # A tibble: 100 × 152
#> fish_1 fish_2 fish_3 fish_4 fish_5 fish_6 fish_7 fish_8 fish_9 fish_10
#> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 47 11 114 11 11 11 28 47 11 21
#> 2 11 47 114 85 28 85 47 85 21 114
#> 3 28 11 180 85 47 11 85 114 85 154
#> 4 11 85 11 85 47 28 180 85 85 154
#> 5 11 85 47 85 85 11 47 180 47 28
#> 6 11 47 180 11 28 11 114 114 85 47
#> 7 28 11 114 85 33 85 114 47 85 28
#> 8 85 114 47 47 85 11 47 154 33 35
#> 9 11 11 47 11 47 33 85 114 47 28
#> 10 85 11 114 85 47 47 114 11 47 47
#> # ℹ 90 more rows
#> # ℹ 142 more variables: fish_11 <dbl>, fish_12 <dbl>, fish_13 <dbl>,
#> # fish_14 <dbl>, fish_15 <dbl>, fish_16 <dbl>, fish_17 <dbl>, fish_18 <dbl>,
#> # fish_19 <dbl>, fish_20 <dbl>, fish_21 <dbl>, fish_22 <dbl>, fish_23 <dbl>,
#> # fish_24 <dbl>, fish_25 <dbl>, fish_26 <dbl>, fish_27 <dbl>, fish_28 <dbl>,
#> # fish_29 <dbl>, fish_30 <dbl>, fish_31 <dbl>, fish_32 <dbl>, fish_33 <dbl>,
#> # fish_34 <dbl>, fish_35 <dbl>, fish_36 <dbl>, fish_37 <dbl>, …Individual identity output in this format may not be very useful for
most users. So, Ms.GSI has a function
indiv_assign() that summarize the reporting group
assignment probabilities for each individual in the mixture.
indiv_assign(mdl_out = msgsi_out, mdl_dat = msgsi_dat)
#> # A tibble: 150 × 13
#> ID Russia `Coastal West Alaska` `North Alaska Peninsula`
#> * <chr> <dbl> <dbl> <dbl>
#> 1 fish_1 0 0.25 0
#> 2 fish_2 0 0.09 0.03
#> 3 fish_3 0.02 0.01 0.27
#> 4 fish_4 0 0.28 0
#> 5 fish_5 0 0.18 0
#> 6 fish_6 0 0.24 0
#> 7 fish_7 0.05 0.06 0.08
#> 8 fish_8 0.19 0.13 0.06
#> 9 fish_9 0.01 0.25 0
#> 10 fish_10 0.04 0.12 0.02
#> # ℹ 140 more rows
#> # ℹ 9 more variables: `Northwest Gulf of Alaska` <dbl>, Copper <dbl>,
#> # `Northeast Gulf of Alaska` <dbl>, `Coastal Southeast Alaska` <dbl>,
#> # `British Columbia` <dbl>, `WA/OR/CA` <dbl>, `Lower Yukon` <dbl>,
#> # `Middle Yukon` <dbl>, `Upper Yukon` <dbl>The summary for individual assignment has a column named
ID that identifies each individual in the mixture. The
reporting groups represent the combined grouping of the broad and
regional baselines. Probabilities in each row should sum up to one.
Trace plot
Ms.GSI has a function for you to make trace plot and examine
the mixing of MCMC chains. Don’t forget to include the group information
(as in groups, p2_groups, or
comb_groups) for the trace history that you want to
plot.
tr_plot(mdl_out = msgsi_out, trace_obj = "trace_comb", pop_info = msgsi_out$comb_groups)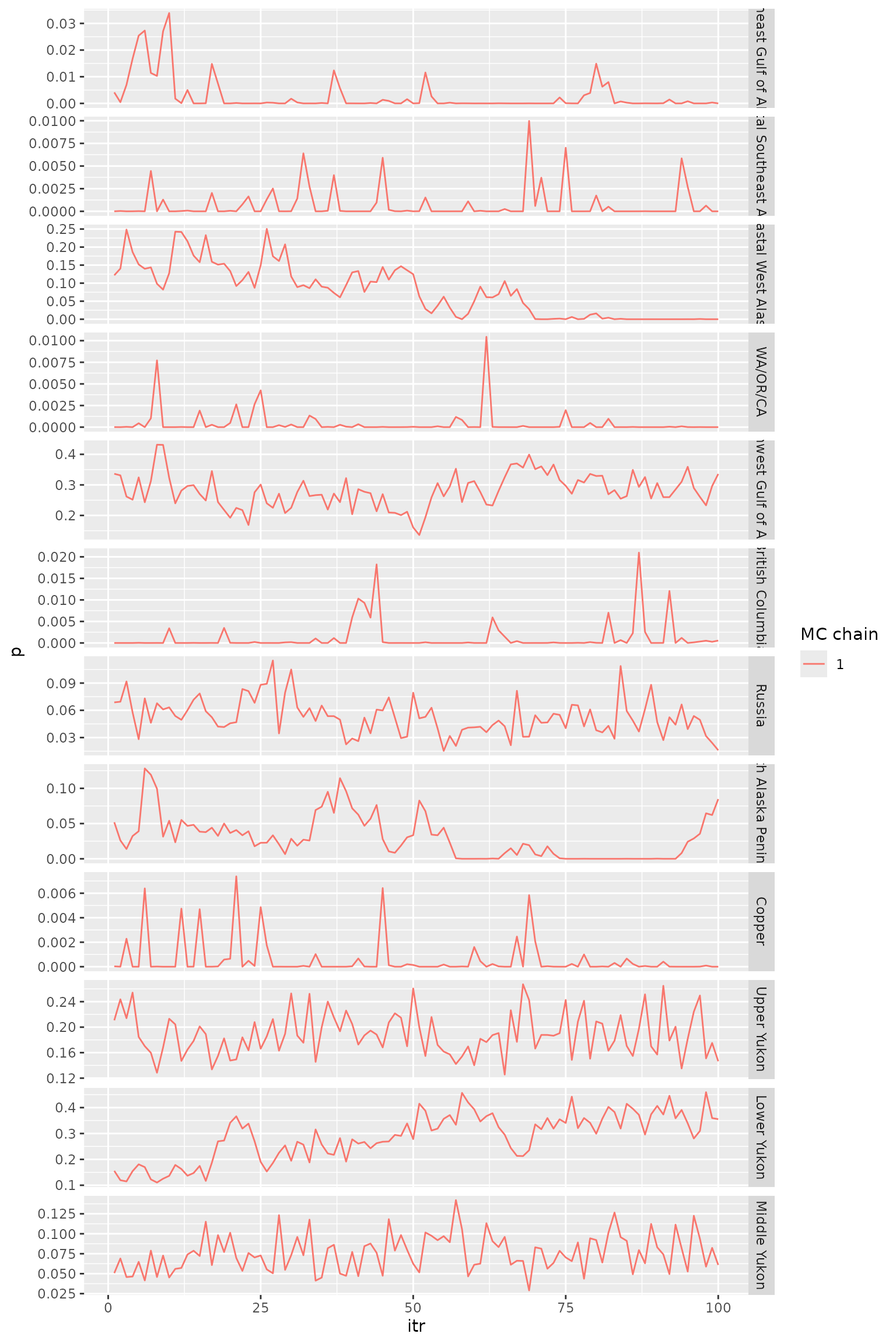
Trace plots for reporting group proportions.
Stratified estimator
stratified_estimator_msgsi() function combines the stock
group estimates of multiple mixtures (i.e., strata) weighted by harvest
numbers or fishing efforts. To do so, you would save the Ms.GSI
output for each mixture in a separate folder under your preferred
directory, then specify the path in the function:
stratified_estimator_rubias(path = ...).
stratified_estimator_msgsi() extracts the stock-specific
total catch/harvest output from the model runs of multiple mixtures and
combine them in the same format of reporting groups or in forms of new
reporting groups by combining old reporting groups or reorganizing
collections. For example, we use the example model output with a new
grouping:
new_groups <- msgsi_out$summ_comb |>
dplyr::select(group) |>
dplyr::mutate(new_repunit = c(rep("Broad", 9), rep("Regional", 3))) |>
dplyr::rename(repunit = group)
new_groups
#> # A tibble: 12 × 2
#> repunit new_repunit
#> <chr> <chr>
#> 1 Russia Broad
#> 2 Coastal West Alaska Broad
#> 3 North Alaska Peninsula Broad
#> 4 Northwest Gulf of Alaska Broad
#> 5 Copper Broad
#> 6 Northeast Gulf of Alaska Broad
#> 7 Coastal Southeast Alaska Broad
#> 8 British Columbia Broad
#> 9 WA/OR/CA Broad
#> 10 Lower Yukon Regional
#> 11 Middle Yukon Regional
#> 12 Upper Yukon Regional
stratified_estimator_msgsi(mdl_out = msgsi_out, mixvec = "Bering example",
new_pop_info = new_groups, new_pop_by = "repunit")
#> # A tibble: 2 × 15
#> repunit mean_sstc sd_sstc median_sstc ci05_sstc ci95_sstc mean sd median
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 Broad 224. 49.5 215 164. 324. 0.448 0.0953 0.431
#> 2 Regional 277. 51.2 278 189. 353. 0.552 0.0953 0.569
#> # ℹ 6 more variables: ci05 <dbl>, ci95 <dbl>, `P=0` <dbl>, `Z=0` <dbl>,
#> # GR <lgl>, n_eff <dbl>The same can be done by specifying collections:
new_groups_collection <- msgsi_out$comb_groups |>
dplyr::left_join(new_groups, by = "repunit")
new_groups_collection
#> # A tibble: 236 × 3
#> collection repunit new_repunit
#> <chr> <chr> <chr>
#> 1 CHBIG92.KIBIG93.KBIGB04.KBIGB95 Northeast Gulf of Alaska Broad
#> 2 CHCRY92.KICRY94.KCRYA05 Coastal Southeast Alaska Broad
#> 3 CHDMT92.KDEER94 Coastal Southeast Alaska Broad
#> 4 CHKAN92.KIKAN93.KKANE05 Coastal West Alaska Broad
#> 5 CHKOG92.KIKOG93.KKOGR05 Coastal West Alaska Broad
#> 6 CHNUU92.KINUS93 Coastal West Alaska Broad
#> 7 CHTAH92.KTAHI04 Northeast Gulf of Alaska Broad
#> 8 CHWHI92.KWHIT98.KWHITC05 Coastal Southeast Alaska Broad
#> 9 KALSE04 WA/OR/CA Broad
#> 10 KANCH06.KANCH10 Northwest Gulf of Alaska Broad
#> # ℹ 226 more rows
stratified_estimator_msgsi(mdl_out = msgsi_out, mixvec = "Bering example",
new_pop_info = new_groups_collection, new_pop_by = "collection")
#> # A tibble: 2 × 15
#> repunit mean_sstc sd_sstc median_sstc ci05_sstc ci95_sstc mean sd median
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 Broad 224. 49.5 215 164. 324. 0.448 0.0953 0.431
#> 2 Regional 277. 51.2 278 189. 353. 0.552 0.0953 0.569
#> # ℹ 6 more variables: ci05 <dbl>, ci95 <dbl>, `P=0` <dbl>, `Z=0` <dbl>,
#> # GR <lgl>, n_eff <dbl>And for those prefer doing things the old way (by multiplying fishing effort by the stock proportions):
stratified_estimator_msgsi(mdl_out = msgsi_out, mixvec = "Bering example",
new_pop_info = new_groups, new_pop_by = "repunit",
naive = TRUE, catchvec = 500, cv = 0.05)
#> # A tibble: 2 × 15
#> repunit mean_harv sd_harv median_harv ci05_harv ci95_harv mean sd median
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 Broad 224. 50.5 211. 161. 325. 0.448 0.0948 0.424
#> 2 Regional 275. 48.4 279. 194. 346. 0.552 0.0948 0.576
#> # ℹ 6 more variables: ci05 <dbl>, ci95 <dbl>, `P=0` <dbl>, `Z=0` <dbl>,
#> # GR <lgl>, n_eff <dbl>Methods (math!)
Pella-Masuda Model
This integrated multistage GSI model is essentially two Bayesian GSI models stacked on top of each other; hence the name “multistage.” The Pella-Masuda model (Pella & Masuda 2001) is the Bayesian GSI model that make up each stage in the integrated multistage model. We will first describe the Pella-Masuda model before discussing the development of a integrated multistage model.
In a group of mixed populations, Pella-Masuda model assigns population identities to each individual based on its genetic make-up (e.g. genotype). Then the model estimates the overall population proportions based on the numbers of individuals assigned to each population. In the fishery context, genetic data of the individuals is called the mixture sample because it consists multi-locus genotype of individual fish collected from a mixed-stock fishery. denotes the mixture sample. In this document, a bold-font letter represents a number set, or a collection of distinct elements. For example, is a set that contains individual elements. And is the count of allele in locus for individual fish , where , , and depends on locus .
Genetic data of the populations is called the baseline sample because it consists genotype compositions of various baseline populations collected at their spawning locations. Researchers select sampling locations to best represent the populations in an area. denotes the baseline sample. is the count of allele in locus for a sample of size collected from baseline population , where .
For both mixture and baseline samples, it is assumed that allele counts in each locus follow a multinomial distribution3. Using another made-up example, in a baseline sample, there are two allele types in locus 1 for population 2. Counts for the two alleles are , and they follow a multinomial distribution with parameters and size . Note that are the relative frequencies of the two alleles in locus 1 for population 2. In a Bayesian framework, we need to specify prior distributions for parameters; therefore, we place a Dirichlet4 prior distribution on with hyperparameters5 . Usually we set the priors to be equal for all loci. In this example, let based on the number of alleles for locus 1.
represents and , together with allele frequencies of other loci and other populations. As you can see, and have the same dimension because each relative frequency corresponds to an allele count. In the model, allele frequencies of baseline populations, , determine population proportions. And population proportions determines the identities of individual fish. Individual identities are then tallied and summarized to update baseline allele frequencies. can be expressed as follows:
Prior distribution for :
,
where
For mixture sample, allele counts in each locus of individual fish also follows a multinomial distributions. If a fish came from a certain population, its distribution of allele counts should resemble the allele frequencies of the baseline population which it came from. However, the identity of the individual fish is unknown so it needs to be estimated. Here we let represent the population identify for the th mixture individual. is composed of 0’s and an 1 with a length (e.g. number of baseline populations). if individual belongs to population , and otherwise. In a made-up example, means that there are five baseline populations, and individual fish #100 comes from population 3.
We place a multinomial prior on with size 1 and probabilities equal to population proportions . We specify a Dirichlet prior distribution on with hyperparameters , where . We usually set to be equal for all reporting groups, but they can be set based on prior knowledge in population proportions. We express as follows:
Prior distribution for :
,
where
As mentioned, for mixture sample, allele counts in each locus of individual fish follows a multinomial distributions. The parameters are allele frequencies of the corresponding baseline population with size the numbers of ploidy for each respective locus. Remember that population identity if individual belongs to population , and otherwise. When multiplying population identities, , and allele frequencies of baseline populations, , only allele frequencies of baseline population which individual belong to would remain while the rest goes to zero. is expressed below. denotes ploidy for each locus.
Moran and Anderson (2018) implement a genetic mixture analysis as a R package, rubias. Their program has been widely used by researchers around the world, including here at the GCL. rubias utilizes a model structure called the conditional genetic stock identification model, or the conditional GSI model, that is modified from the Pella-Masuda model. The main difference between the two models is that, in the conditional model, is integrated out of the distribution of mixture sample, . That is, baseline allele frequencies are not being updated in the model. The result of that, takes a form of a compound Dirichlet-multinomial distribution (Johnson at el. 1997):
,
where is . We are not going to attempt proving the theory behind the conditional model in this document (details can be found in Moran & Anderson 2018). But since has been integrated out of , the process for estimating parameters is simpler and more streamlined. We have implemented conditional GSI in each stage of our integrated multistage model.
Extend to multistage
In a multistage setting, we refer to a baseline that covers the whole range of a mixed stock fishery as a broad-scale baseline. The broad-scale baseline typically covers a wide range of geographic areas but does not have a comprehensive collection of reference populations nor genetic markers to resolve differences between local populations within a sub-region. These smaller sub-regions of a broad-scale baseline are covered by regional baselines with higher resolutions. We generalize the conditions of the Ms.GSI model to allow multiple regional baselines to be included, although we programmed Ms.GSI to deal with only one regional baseline vs. one broad-scale baseline.
Let there be populations in the broad-scale baseline and indexed as . Each of these broad-scale populations may belong to exactly 0 or 1 sub-region for which regional baselines might be available. These regional baselines have different sets of genetic markers than the broad-scale baseline and typically include additional populations that are not represented in the broad-scale baseline. Allow for there to be disjoint sub-regions indexed by , with each sub-region represented by a distinctive regional baseline. We employ a superscript upon variables to indicate a quantity associated with regional baseline . Populations in different sub-regions cannot overlap, and each population only occurs once among the regional baselines. Let index the populations within these regional baselines and denotes the number of populations within regional baseline .
In the Ms.GSI framework, the two stages are connected because the regional group membership of an individual is conditional on whether the broad-scale group membership of that individual belongs to the area of that particular sub-region. The following describes the conditional relationship between the broad-scale and the regional baselines:
,
where and are vectors of indicators ( or ) identifying the broad-scale and regional populations that individual belongs to. denotes the broad-scale populations that belong to the areas represented by the reporting groups of region , and is a vector of all zeros.
Ultimately, we want to estimate the fraction of individuals in the mixture that come from each of the sub-regional populations, as well as from any of the populations in the broad-scale baseline that are not associated with a regional baseline. denotes the mixture proportion of the th population in region , and denotes the mixture proportion of population in the broad-scale baseline. Thus, we endeavor to estimate the mixture proportions for each such that and along with , where , with denoting the broad-scale populations that do not belong to any areas represented by regional baselines. Lastly, we multiply by for each region , so the scaled for all regions and , where would sum to one.
Gibbs Sampler: where the fun go round and round
Deriving the values of parameters in each stage of the integrated multistage model requires finding the joint posterior distribution of Pella-Masuda model in each stage, . In this section, we will introduce the concepts and algorithm to sample from this posterior distribution in a single baseline Pella-Masuda model, which then can be extend to an integrated multistage framework.
Gibbs sampler is a type of MCMC methods that sequentially sample parameter values from a Markov chain. With enough sampling, the Markov chain will eventually converge to the desire distribution of interest. The most appealing quality of Gibbs sampler is its reduction of a multivariate problem (such as Pella-Masuda model) to a series of more manageable lower-dimensional problems. A full description of Gibbs sampler and MCMC methods is beyond the scope of this document; however, further information can be found in numerous resources devoting to Bayesian data analysis (see Carlin & Louis 2009; Robert & Casella 2010; Gelman et al. 2014)
To illustrate, suppose we would like to determine the joint posterior distribution of interest, , where . Most likely the multivariate would be too complicated to sample from. However, if we can figure out how to break up the joint posterior distribution into individual full conditional distributions, each parameter in can be sampled one by one sequentially using a Gibbs sampler algorithm. The process begins with an arbitrary set of starting values and proceeds as follows:
For , repeat
Draw from
-
Draw from
⋮
- Draw from
This would work best if the full conditionals are some known distributions that we can easily sample from (although it’s not required). In our case with Pella-Masuda model, we rely on two main concepts, the Bayes theorem and conjugacy, to do the trick. Briefly, for estimating parameters from data , according to Bayes Rule, . is the joint posterior distribution for parameters , is the likelihood of observing the data given the parameters, is the prior distribution of the parameters, and is the constant marginal distribution of the data. is often mathematically difficult to obtain; however, because is a constant number, we can ignore it by reducing the posterior distribution to .
So, how does Bayes Rule help us estimating parameters in the Pella-Masuda model? First, the joint posterior distribution has to be split up into smaller pieces. That is, we separate the joint posterior into likelihood of the data and priors for the parameters:
With some re-arrangements and hand-waving, we arrive at the full conditional distributions for and :
Next, we take advantage of a mathematical property called conjugacy to help us determine the conditional distributions. Based on this property, the posterior distribution follows the same parametric form as the prior distribution when prior is a conjugate family for the likelihood. For example, if the likelihood of data is binomial distribution and the prior of parameter is beta distribution, then the posterior is also beta distribution because beta is a conjugate family for binomial. There are many conjugate families, and Dirichlet and multinomial are another example.
Utilizing conjugacy property, we will determine each of the full conditional distributions for and .
Conditional distribution p(q|x, y, z, )
We determine that is Dirichlet-distributed because Dirichlet prior is a conjugate family for the multinomial likelihoods and . To determine the exact parameterization for the posterior distribution, we need to derive the prior and likelihoods first.
Likelihood can be derived in two steps. The first step we conditioned the likelihood on so that
,
where is the relative frequency of multi-locus genotype for individual in population . In the next step, we derive :
Then we combine the two,
Deriving likelihood is more straightforward. It is the product of relative frequency of multi-locus genotype for each population:
And is Dirichlet prior distribution. Its probability density has a kernel6 of . We can express the likelihood as
.
Put all the likelihoods together,
It is elementary to recognize that is the kernel for Dirichlet distribution. Hence,
.
Conditional distribution p(p|z, )
Using the same logic as previously, is also Dirichlet-distributed due to a Dirichlet prior and a multinomial likelihood .
Once again, we recognize it as the kernel for Dirichlet distribution:
Algorithm
There is one more distribution to figure out before we can start our Gibbs sampler routine (and you thought we’re all set, lol). We would need to know how to sample , the population identity for individual fish (in components 1 and 2) given the population proportions and genotype. If the probability of fish belong to population is , and the likelihood of observing relative frequency of genotype for fish in population is , then the probability of fish belong to population given the population proportions and genotype is . The denominator should sum to one, so we only need to calculate the numerator.
has the following distribution:
,
where . We draw the initial values for based on its prior distribution.
Once we figured out all the pieces in the Gibbs sampler for the single baseline framework, we can extend the concept to a multistage framework. Conceptually, we use a Gibbs sampler to sample from the full conditionals of , , , , , and . The full conditional distributions of the broad-scale baseline in the multistage framework stay the same as their counterparts in a single baseline framework. The full conditional distributions for the Gibbs sampler at the regional stage are:
,
,
and ,
where .
We initiate the Gibbs sampler with starting values for , , and based on their prior distributions. We use subscript to denote th iteration of the Gibbs sampler. Sampling for the fully Bayesian model proceeds as follows:
For , repeat
Determine the group memberships of mixture individuals at the broad-scale stage, .
Determine the group memberships of mixture individuals for each sub-region at the regional stage, , .
Draw updated values, , , and from , , and respectively.
should be large enough to ensure the simulations converge to the posterior distribution. Usually it takes thousands of iterations. Implementing the conditional GSI model only requires a slight modification from the above algorithm. and only need to be calculated once at the initial step without further updates, otherwise the procedures remain the same.
References
Brooks, S. P., and A. Gelman. 1998. General methods for monitoring convergence of iterative simulations. Journal of Computational and Graphical Statistics. 7:434–455.
Carlin, B. and T. Louis. 2009. Bayesian Methods for Data Analysis, 3rd Edition. CRC Press. New York.
Gelman, A., and D. B. Rubin. 1992. Inference from iterative simulation using multiple sequences. Statistical Science. 7:457–472.
Gelman, A., J. Carlin, H. Stern, D. Dunson, A. Vehtari and D. Rubin. 2014. Bayesian Data Analysis, 3rd Edition. CRC Press. New York.
Johnson, N.L., Kotz, S., and Balakrishnan, N. 1997. Discrete multivariate distributions. Wiley & Sons, New York.
Lee. E., T. Dann, and H. Hoyt. 2021. Yukon River Chinook Genetic Baseline Improvements. Yukon River Panel Restoration and Enhancement Fund Final Report, URE-163-19N.
Moran, B.M. and E.C. Anderson. 2018. Bayesian inference from the conditional genetic stock identification model. Canadian Journal of Fisheries and Aquatic Sciences. 76(4):551-560. https://doi.org/10.1139/cjfas-2018-0016
Pella, J. and M. Masuda. 2001. Bayesian methods for analysis of stock mixtures from genetic characters. Fish. Bull. 99:151–167.
Robert, C. and G. Casella. 2010. Introducing Monte Carlo Methods with R. Springer. New York.
Templin, W. D., J. E. Seeb, J. R. Jasper, A. W. Barclay, L. W. Seeb. 2011. Genetic differentiation of Alaska Chinook salmon: the missing link for migratory studies. Mol Ecol Resour. 11(Suppl 1):226-246. doi:10.1111/j.1755-0998.2010.02968.x.